|
|
|
| |
Se agrupa bajo esta denominación un alto número de enfermedades
heterogéneas que tienen en común comprometer predominantemente
el intersticio pulmonar en forma difusa, ya sea de partida o
como consecuencia de focos sucesivos. La conformación
del grupo es un tanto discrecional ya que algunas tienen una
fisonomía o etiología suficientemente específica,
suelen ser clasificadas como entidades aparte o en otros grupos
(neumonitis intersticiales infecciosas, tuberculosis miliar,
linfangiosis carcinomatosa, edema pulmonar, neumoconiosis, asbestosis,
etc.).
Las enfermedades intersticiales son entidades agudas, subagudas o crónicas
derivadas de combinaciones de diferentes tipos y grados de infiltración
inflamatoria y de fibrosis del intersticio, a lo que se agregan,
en grado también variable, aliteraciones del epitelio
alveolar y relleno del espacio alveolar. Parte importante de
estas condiciones progresa a una remodelación profunda
del pulmón, con grave deterioro funcional. Como su diferenciación
no es siempre fácil y presentan un cuadro clínico,
radiológico y fisiológico similar, durante parte
importante de su proceso diagnóstico se abordan como
un síndrome. El hecho de que enfermedades muy diferentes
den un cuadro clínico similar se explica por la existencia
de algunos mecanismos en común en su patogenia y por
la inespecifidad con que el pulmón responde ante diferentes
agresores.
En general, el enfrentamiento de estas enfermedades es difícil y necesita
del trabajo conjunto de varias especialidades, pero la detección
inicial del síndrome es responsabilidad del clínico
general, a quien también corresponderá el manejo
de algunas de las entidades de causa conocida. Por ello veremos
primero las características del síndrome para
luego abordar algunas de las entidades más frecuentes
de causa conocida, y finalmente entregar una información
sumaria sobre las principales enfermedades que deben referirse
al especialista.
PATOGENIA
Las enfermedades intersticiales difusas pueden estar confinadas exclusivamente
al pulmón o ser parte de una patología sistémica
multiorgánica (enfermedades colágenas, enfermedades
crónicas intestinales, hepáticas o renales),
a las cuales pueden, ocasionalmente, preceder. En ambas situaciones
se combinan en grado variable tres mecanismos básicos:
daño alveolar difuso, reacción inflamatoria
y activación fibroblástica, que conducen a diferentes
formas de reparación, remodelación o destrucción.
(Figura 39-1).
Figura 39-1. Patogenia de las enfermedades alveolares difusas.
La lesión inicial produce un daño alveolar
que inicia una reacción inflamatoria o una activación
fibro-proliferativa. En algunos casos ambos procesos van
a la reparación, con o sin secuelas. En otros, los
propios mediadores inflamatorios causan más daño
alveolar, con la consiguiente formación de un círculo
vicioso que perpetúa el trastorno.
DAÑO ALVEOLAR DIFUSO.
El agente causal, conocido o desconocido, llega a la pared alveolar por vía
inhalatoria o por vía sanguínea. En contacto
con el tejido pulmonar, produce un daño en sus células
parenquimatosas y en el estroma, cuya extensión e intensidad
es muy variable. Ordinariamente es enmascarado por las alteraciones
que siguen.
REACCION INFLAMATORIA
La lesión inicial es seguida de una fase reactiva de variada intensidad
y naturaleza. Aparecen células inflamatorias, que infiltran
el intersticio y pueden ocupar el alvéolo, provenientes
de la transformación y activación de células
tisulares (histiocitos, plasmocitos, linfocitos) y de la atracción
quimiotáctica de células circulantes (neutrófilos,
eosinófilos, linfocitos, etc.). El fenómeno
inflamatorio puede ser autolimitado o seguir progresando,
por persistencia del agente causal o por efecto de las enzimas
y los mediadores liberados por las propias células
inflamatorias, que incrementan el daño alveolar. Esta
nueva destrucción provoca mayor reacción inflamatoria,
originando un círculo vicioso que explica la progresión
observada en muchos casos después de que la noxa causal
habría dejado de actuar. En algunos casos el tejido
pulmonar inicialmente alterado puede no ser reconocido por
el organismo y comportarse como un antígeno que provoca
la formación de auto-anticuerpos que aumentan el daño
y mantienen la inflamación. La multiplicidad de agentes
causales, vías de contacto y diferencias individuales
de reactividad explica la amplia gama de variantes que pueden
presentarse.
El carácter difuso del proceso puede deberse tanto a que la noxa afecta
de partida a todo o a la mayor parte del pulmón como
a la suma de focos sucesivos. El primer mecanismo conduce
a lesiones histológicas homogéneas, mientras
que el segundo lleva a la coexistencia de zonas normales con
otras en diferentes etapas de reacción y reparación.
La evolución temporal de los procesos descritos puede
también variar ampliamente desde formas agudas, que
en semanas mejoran, se detienen o llevan a la muerte, a formas
crónicas de hasta 15 o más años de evolución,
con todas las formas intermedias posibles.
ACTIVACION FIBROBLASTICA
La proliferación y activación fibroblástica aparece en algunos
casos como una consecuencia de la inflamación y en
otros sería un fenómeno primario con focos fibroblásticos,
independientemente del grado previo de inflamación.
En ambos casos el resultado es una fibrosis progresiva. Los
efectos de este fenómeno son también variados:
cuando la membrana basal del epitelio alveolar queda relativamente
indemne, se puede producir una reparación que puede
ser íntegra o con daño funcional de nula o leve
expresión clínica. En el otro extremo, si la
fibrosis es intensa y extensa los neumocitos tipo II proliferados
engloban células y detritus intraalveolares y los incorporan
a los tabiques alveolares, con lo que la arquitectura resulta
fuertemente distorsionada.
NOMENCLATURA
Aunque las enfermedades intersticiales difusas de causa conocida forman
parte del diagnóstico diferencial inicial de síndrome
, una vez que se identifican y adquieren un nombre,
generalmente no se siguen incluyendo en el grupo sino que
se manejan individualmente.
En cambio, la mayoría de las condiciones de etiología desconocida
siguen considerándose como parte de grupos o subgrupos
y existe una multiplicidad de denominaciones de variable aceptación
Como grupo son a veces denominadas "enfermedades infiltrativas
difusas", " enfermedades intersticiales difusas"o
"enfermedades parenquimatosas difusas" y sus subdivisiones
son muy variadas y diferentes. La nomenclatura más
clara y que goza de mayor consenso es la propuesta por las
sociedades Americana de Tórax y Respiratoria Europea,
que se resume en la Tabla 39-1.
Tabla 39-1
ENFERMEDADES PARENQUIMATOSAS DIFUSAS DEL PULMON
Clasificación simplificada de las Sociedades Americana
de Tórax y Respiratoria Europea
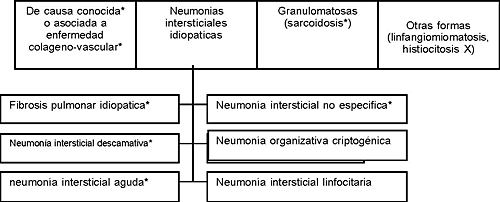
Por el momento no es necesario profundizar mayormente en esta clasificación,
cuya significación e importancia se aclararán
cuando se analice algunos de sus principales integrantes,
que están marcados con un asterisco.
ANATOMIA PATOLOGICA
Se han descrito algunos patrones histológicos definidos, pero, por no
ser exclusivos o específicos de una sola entidad clínico-patológica,
no siempre permiten por sí solos fundamentar el diagnóstico
final. Este sólo puede plantearse a partir del análisis
conjunto de la histología con los datos aportados por
el clínico y por el radiólogo, siendo obvio
que la visión morfológica aislada de una biopsia,
a pesar de su alto valor, no puede decir si existen factores
causales clínicamente identificables o se trata de
una forma idiopática o criptogénica; si la enfermedad
está confinada al pulmón o es parte de una enfermedad
sistémica y cuál es la distribución y
extensión de las lesiones en ambos pulmones.
Los diferentes patrones incluyen combinaciones variables de:
-
Efectos directos de la lesión, representados por daño del epitelio
alveolar o alteraciones de la pared capilar. Cuando el agente
causal es microbiano o particulado, puede constatarse su
presencia en el examen histológico o se pueden detectar
embolias cruóricas, agregados plaquetarios, lípidos,
etc. Usualmente los procesos reaccionales ocultan esta fase
inicial de la afección.
-
Inflamación que se desarrolla preponderantemente en el intersticio , pero
pueden extenderse a los espacios alveolares y comprometer
bronquíolos y vasos pulmonares. Los tipos de células
inflamatorias varían según la entidad patológica
y pueden disponerse en forma difusa o formar granulomas.
Usualmente hay edema y en algunas pueden encontrarse membranas
hialinas formadas por exudados proteináceos y fibrina.
-
Proliferación fibroblástica que puede ser consecuencia de la inflamación
previa o presentarse como focos fibroblasticos con escasa
inflamación. En ambos casos se asiste a grados variables
de fibrosis que, en los casos extremos oblitera espacios
alveolares y tracciona las paredes de los bronquíolos,
con formación de cavidades quísticas. Este
conjunto constituye la etapa terminal, llamada panal de
abejas, con pérdida total de la capacidad funcional.
Entre ambos extremos es posible encontrar todo tipo de combinaciones
intermedias.
-
Diversos grados de hiperplasia e hipertrofia de fibras musculares lisas.
No existe un cuadro fisiopatológico único, pero hay algunos elementos
comunes suficientemente frecuentes como para delinear una
fisonomía básica característica. Variando
de acuerdo a la extensión, duración e intensidad
de la afección, las principales alteraciones funcionales
producidas por la infiltración intersticial difusa
son:
Disminución de la distensibilidad pulmonar:debida
tanto a un aumento de la rigidez del intersticio como a la
obliteración de alvéolos. La necesidad de generar
mayores presiones negativas para ventilar el pulmón
significa un aumento del trabajo respiratorio, que explica
la disnea de estos pacientes. Además, hay un aumento
de reflejos propioceptivos que se traduce por taquipnea con
reducción del volumen corriente. Las alteraciones de
la distensibilidad también determinan una restricción,
frecuentemente progresiva, de los volúmenes pulmonares,
con relación VEF1 /CVF normal o aumentada. Generalmente
no hay elementos obstructivos, pero en algunas afecciones
se comprometen también los bronquios finos. La tos
se debería también a la mayor rigidez con estimulación
de receptores propioceptivos.
Hipoxemia: se debe a múltiples mecanismos. Por
una parte las áreas menos distensibles reciben menos
ventilación, con disminución regional de la
relación  /Q que es la alteración preponderante. Por otra,
áreas obliteradas, colapsadas o con relleno alveolar
actúan como cortocircuitos y, finalmente, existen trastornos
de difusión debido a la interposición de infiltrado
inflamatorio y/o fibrosis entre alvéolos y capilares.
En la medida que estas alteraciones se extienden, se produce
primero un aumento de la diferencia alvéolo-arterial
de O2 en ejercicio y
luego en reposo, apareciendo más adelante hipoxemia.
Por la alta difusibilidad del CO2
y la eficacia de la hiperventilación compensatoria
para su remoción, la retención de este gas solo
se presenta en etapas terminales.
/Q que es la alteración preponderante. Por otra,
áreas obliteradas, colapsadas o con relleno alveolar
actúan como cortocircuitos y, finalmente, existen trastornos
de difusión debido a la interposición de infiltrado
inflamatorio y/o fibrosis entre alvéolos y capilares.
En la medida que estas alteraciones se extienden, se produce
primero un aumento de la diferencia alvéolo-arterial
de O2 en ejercicio y
luego en reposo, apareciendo más adelante hipoxemia.
Por la alta difusibilidad del CO2
y la eficacia de la hiperventilación compensatoria
para su remoción, la retención de este gas solo
se presenta en etapas terminales.
Hipertensión pulmonar y corazón pulmonar, que se desarrollan
por compromiso anatómico de los vasos pulmonares y
por vasoconstricción hipoxémica. Se exteriorizan
primero en ejercicio para establecerse en reposo en los casos
extensos y avanzados.
ETIOLOGIA
Los agentes y mecanismos capaces de causar compromiso pulmonar intersticial difuso
son numerosos y se enumeran en la Tabla 39-2.
| TABLA
39-2
AGENTES Y MECANISMOS CAUSANTES O ASOCIADOS A COMPROMISO
INTESTICIAL DIFUSO |
| 1.-INFECCIONES
-Bacterias: Tuberculosis miliar
-Virus: Planteados como precursores en algunas formas
de causa desconocida.
-Hongos: Neumocistes jiroveci, aspergilio,
histoplasma, coccidioides etc.
2.-AGENTES FISICOQUIMICOS
-Inhalatorios:
-Partículas: Neumoconiosis
(silicosis, asbestosis, siderosis, etc.).
-Gases: NO2, SO2, amoníaco,
cloro, oxígeno en concentración alta por
tiempo prolongado
-Por aspiraciones recurrentes:
-Contenido gástrico (reflujo
gastroesofágico)
-Lípidos
-Transtorácicos:
-Radiación
-Hematógenos:
-Tóxicos: paraquat (matamalezas)
- Drogas: por acción tóxica
(Tabla 41-3)
3.-NEOPLASIAS MALIGNAS
-Cáncer bronquíolo-alveolar
-Linfangiosis carcinomatosa
-Enfermedades linfoproliferativas
-Leucemias
4.-GENETICAS O FAMILIARES
-Enfermedad de Gaucher, Niemann-Peak, esclerosis
tuberosa, etc.
5.-METABOLICAS
-Uremia
6.-CIRCULATORIAS
Embolias:
-Tromboembolismo múltiple
-Embolia grasa
-Embolia de cuerpo extraño en drogadictos
(talco)
Hemodinámicas:
-Edema crónico por hipertensión
pulmonar postcapilar: estenosis mitral, insuficiencia
cardíaca congestiva, enfermedad veno-oclusiva
7.-INMUNOLOGICAS
Neumonitis por hipersensibilidad:
-Alveolitis alérgica extrínseca
por hongos, proteínas aviarias, enzimos, etc.
(Tabla 41-2).
-Reacción alérgica a drogas (Tabla
41-3)
Enfermedades sistémicas o colagenopatías:
-Esclerosis sistémica progresiva, enfermedad
reumatoidea, dermatomiositis, lupus eritematoso
diseminado, etc.
8.-DE CAUSAS O MECANISMOS DESCONOCIDOS
Neumonías intersticiales idiopáticas
Fibrosis pulmonar idiopática
Neumonía intersticial inespecífica
Neumonía intersticial aguda
Enfermedad pulmonar intersticial asociada a bronquiolitis
respiratoria.
Neumonía intersticial descamativa
Neumonía organizativa criptogénica (COP)
Neumonía intersticial linfocítica
Sarcoidosis.
Granulomatosis de Wegener
Histiocitosis X o histiocitosis de células de
Langerhans
Hemosiderosis idiopática
Síndrome de Goodpasture
Proteinosis alveolar
Linfangiomiomatosis |
En esta tabla conviene resaltar lo siguiente:
- La lista es muy amplia en cuanto a factores causales o asociados, por lo que
incluye muchas afecciones que, por tener una fisonomía
muy precisa, no siempre se clasifican como parte de este
grupo, aunque pueden plantearse como diagnóstico
diferencial. Por otra parte la lista no es exhaustiva,
ya que las enfermedades de este grupo sobrepasan las 130
y están en constante modificación y crecimiento.
- Las categorías empleadas no son excluyentes, puesto que algunas se definen
por el agente etiológico y otras por el mecanismo
patogénico, elementos que pueden sobreponerse.
El objetivo básico de esta enumeración es ilustrar la multiplicidad
de caminos que pueden llevar a un resultado similar. No
tiene objeto memorizar los detalles, sino tener presentes
las grandes categorías y estar preparado para detectar
oportunamente una síndrome infiltrativo difuso e
iniciar su estudio diagnóstico básico.
CARACTERISTICAS CLINICAS
El síndrome sugerente de compromiso alveolar infiltrativo difuso está
constituido por tres elementos básicos:
-
Disnea de esfuerzos, generalmente progresiva con o sin tos seca.
-
Compromiso radiográfico retículo-nodular difuso, con o sin elementos
de relleno alveolar
-
Limitación ventilatoria restrictiva, con hipoxemia que se acentúa
o aparece en ejercicio
En etapas iniciales, algunas de estas alteraciones pueden no estar presentes.
La disnea es primero de grandes esfuerzos, avanzando paulatinamente
para presentarse con esfuerzos cada vez menores, hasta llegar
finalmente a ser de reposo. En las formas agudas la progresión
puede ser extremadamente rápida, mientras que en las
formas crónicas es lenta, aunque puede presentar agravaciones
espontáneas bruscas. Es habitual que, debido a la estimulación
de receptores en el parénquima rígido, la disnea
se acompañe de tos seca irritativa que puede llegar
a ser extremadamente molesta.
Aunque la radiografía de tórax puede ser normal en fases tempranas,
la imagen más frecuente y sugerente es la de tipo retículo-nodular,
con grados variables de relleno alveolar y reducción
del volumen pulmonar. La tomografía axial computarizada
(TAC) es considerablemente más sensible que la radiografía
y con alguna frecuencia sus imágenes son suficientemente
específicas como para sugerir alguna etiología
o entidad definida, siempre que sea de buena calidad y analizada
por un radiólogo experto.
En la espirometría se observa característicamente una limitación
restrictiva, aunque inicialmente puede estar "dentro de límites
normales" debido al amplio rango de éstos. En los gases
arteriales, lo más precoz es el aumento de la diferencia
alvéolo arterial detectable primero en ejercicio y,
más adelante, la caída de la PaO2.
La medición de estas alteraciones, junto a la prueba
de caminata en 6 minutos, son especialmente importantes para
controlar la evolución de la enfermedad y el efecto
del tratamiento. La capacidad de difusión es otro índice
importante para el diagnóstico y control del tratamiento
especializado.
Otras manifestaciones clínicas que pueden estar presentes, sin que formen
parte obligada del síndrome, son las, crepitaciones
finas de final de inspiración, especialmente en las
partes dependientes del pulmón, y el dedo hipocrático,
que es más frecuente en la fibrosis pulmonar idiopática.
PROCESO DIAGNOSTICO GENERAL EN ENFERMEDADES INTERSTICIALES
Cuando los datos anamnésicos y de examen físico sugieren la posibilidad
de una enfermedad intersticial, los pasos siguientes son una
radiografía de tórax, en busca de una imagen
compatible, una espirometría para objetivar el trastorno
restrictivo y los gases en sangre para detectar el aumento
de diferencia alveólo-arterial o la hipoxemia y su
aumento con el ejercicio. La TAC, que se ha constituido en
una herramienta indispensable para el estudio avanzado de
estas afecciones, puede incorporarse a esta etapa de estudio
básico o dejarse para el estudio por especialista.
Confirmada la existencia de una enfermedad intersticial difusa, debe determinarse
si la lesión es exclusivamente pulmonar o si forma
parte de una enfermedad sistémica (colágenas,
granulomatosis, vasculitis, etc.) o es efecto de patología
de otro órgano (metástasis, embolias, distrés
respiratorio del adulto, etc.). Si es exclusivamente pulmonar
debe descartarse metódicamente la existencia de infecciones,
efectos tóxicos de drogas, exposición a agentes
inhalatorios, etc. (Tablas 39-1, 41-1, 41-2 y 42-1). Es fundamental
evitar que una enfermedad se rotule como idiopática
por efecto de una mala anamnesis.
Todas estas fases del estudio corresponden, en general, al clínico general,
quien deberá decidir si se trata de una enfermedad
que él es capaz de manejar o si debe obtener ayuda
o derivar al paciente.
En la etapa especializada, si no existen antecedentes o elementos clínicos
que apoyen claramente una etiología, será necesario
considerar la TAC de alta resolución, que en manos
expertas puede dar indicios diagnósticos que permiten
decidir un tratamiento.
Si persiste la indefinición se entrará a considerar la biopsia
pulmonar. La justificación de este procedimiento invasivo
reside en el riesgo que significa dejar sin tratamiento una
afección de este tipo, o usar un tratamiento riesgoso
como corticoides o inmunosupresores en enfermos en los cuales
son inútiles. El tejido debe ser obtenido de preferencia
por toracoscopía o por toracotomía, de manera
que se pueda elegir el sitio de la biopsia evitando áreas
de pulmón sano y de pulmón terminal. Las muestras
deben ser preferentemente de más de un lóbulo
y de tamaño suficiente como para poder identificar
la arquitectura histológica general, que es un elemento
definitorio en el diagnóstico diferencial. Además,
es frecuentemente necesario efectuar estudios inmunológicos
y microbiológicos, que exigen contar con tejido suficiente.
Es importante tener presente que el riesgo de este procedimiento
es muy bajo cuando lo efectúa un cirujano experto,
con una evaluación previa adecuada, pero, por ser la
biopsia un método invasivo, sólo debe efectuarse
cuando la información que pueda entregar constituya
un factor de decisión para una conducta concreta. En
otras palabras, no debe hacerse si el resultado es indiferente,
pero no debe demorarse si es necesaria para tomar decisiones.
Si se sospecha una enfermedad infiltrativa de histología homogénea
característica, que permita el diagnóstico con
muestras pequeñas de tejido (tuberculosis miliar, sarcoidosis,
carcinomatosis, proteinosis, etc.) y, sobre todo si ésta
afecta el intersticio axial que acompaña a los bronquios,
puede recurrirse a una biopsia transbronquial endoscópica.
En los últimos años se ha desarrollado la técnica del lavado
broncoalveolar, consistente en la inyección, a través
de un broncoscopio, de 100 a 300 ml de suero fisiológico
en un segmento pulmonar y su recuperación por aspiración.
Con ello se obtiene una muestra de células intraalveolares,
cuya composición sería representativa de lo
que ocurre en los espacios alveolares y posiblemente, en el
intersticio. Si bien este método ha resultado una herramienta
poderosa para la investigación, su aplicación
clínica en esta área está aún
en etapa de definición, por lo que su eventual uso
e interpretación corresponden a centros especializados.
VINCULOS EXTRAPROGRAMATICOS
American
Thoracic Society/European Respiratory Society International
Multidisciplinary Consensus Classification of the Idiopathic
Interstitial Pneumonias Am. J. Respir. Crit. Care Med. 2002
;165: 277-304
|
|
|

