|
|
|
| |
NEUMONIAS INTERSTICIALES IDIOPATICAS
Este subgrupo corresponde netamente al área
especializada, donde constituye un problema difícil por
ser de nomenclatura cambiante, diagnóstico diferencial
complejo y porque la mayor parte de ellas aún carece
de un tratamiento de eficacia demostrada.
Las primeras descripciones de Hamman y Rich
correspondieron a casos subagudos que evolucionaron a la muerte
en pocos meses. Posteriormente fueron descritas formas de evolución
crónica algunas de cuyas etapas evolutivas, fueron consideradas
como enfermedades distintas, dando origen a una frondosa y confusa
nomenclatura. Sólo en los últimos ocho años
se ha asistido a una clarificación conceptual, que aún
está en proceso.
En la medida que se han progresado en el tratamiento
de las enfermedades infecciosas, que la exposición del
hombre a agentes ambientales y drogas ha aumentado y que han
aumentado la expectativa de vida los sujetos de edades mayores
se está observando un incremento de estas afecciones,
que ya no son una rareza (incidencia de 14 a 30/100.000 en EEUU),
de manera que deben tenerse presentes al enfrentarse con el
síndrome intersticial difuso.
Mencionaremos sumariamente las características
básicas de las entidades que actualmente se agrupan bajo
esta denominación.
FIBROSIS PULMONAR IDIOPATICA (Idiopathic pulmonary
fibrosis: IPF)
Este nombre englobaba a varias de las entidades
del grupo pero hoy se reserva exclusivamente para la entidad
más frecuente y grave. Su sustrato histológico
es el patrón llamado UIP (Usual Interstitial Pneumonia),
que se caracteriza por la presencia de focos fibroblásticos,
independientes del grado de inflamación, y por presentar
áreas en diferentes etapas de desarrollo, con predominio
bi-basal y subpleural que conducen finalmente a un extenso
panal de abejas. Es de naturaleza progresiva y carece de un
tratamiento eficaz, con una sobrevida mediana de 2,5 a 3,5
años, aunque hay casos más prolongados. Si el
cuadro clínico es concordante y la TAC muestra la distribución
característica con panal de abejas el diagnóstico
puede hacerse con alta certeza sin necesidad de recurrir a
biopsia. La certeza del diagnóstico es crucial para
evitar un tratamiento corticoidal intenso que tiene un alto
riesgo. Es importante recalcar que el patrón histopatológico
UIP no es exclusivo de esta condición, encontrándose
también en asbestosis y en el compromiso pulmonar de
enfermedades sistémicas como artritis reumatoidea y
esclerosis sistémica progresiva.
NEUMONIA INTERSTICIAL NO ESPECIFICA (Non-specific
interstitial pneumonia: NSIP)
Esta entidad está aún en definición
y se caracteriza por ser histológicamente homogénea,
con inflamación importante y menos tendencia al panal
de abejas. Antes se confundía con la anterior, explicando
los casos de "fibrosis pulmonar idiopática"
que aparecían respondiendo a los corticoides. La importancia
de su identificación reside en esta posibilidad de
curación o control. Su patrón histológico
se puede presentar también en enfermedades colágenas,
daño por drogas, infecciones e hipersensibilidad. Debe
evitarse su confusión con la entidad del mismo nombre
que se presenta en pacientes con SIDA
NEUMONIA INTESTICIAL AGUDA (Acute interstitial pneumonia:
AIP)
Es un cuadro agudo de inflamación
intersticial con insuficiencia respiratoria sin causa aparente,
con una alta mortalidad a los 2 a 3 meses, que correspondería
a los casos inicialmente descritos como síndrome de
Hamman-Rich. Su sustrato histológico es de un daño
alveolar difuso en organización con inflamación
intensa, proliferación fibroblástica y membranas
hialinas.
NEUMONIA INTERSTICIAL DESCAMATIVA (Desquamative intertitial
pneumonia: DIP)
Se observa preferentemente en fumadores y
se caracteriza por un extenso relleno alveolar de macrófagos
pigmentados. Su pronóstico es relativamente bueno,
ya que responde a los corticoides y a la cesación del
tabaco.
NEUMONIA INTERSTICIAL ASOCIADA A BRONQUIOLITIS RESPIRATORIA
Se considera que formaría parte del espectro de la
DIP.en que se produce una inflamación de lo bronquiolos
respiratorios.
NEUMONIA ORGANIZATIVA CRONICA (Chronic organizing pneumonia:
COP)
También conocida como BOOP (Bronchiolitis
obliterans organizing pneumonia), esta entidad es una
reacción histológica inespecífica que
se presenta como un cuadro febril con áreas de consolidación
dispersas de preferencia subpleurales ligadas a relleno alveolar
en organización con o sin bronquiolitis asociada. Puede
ser idiopática o presentarse en enfermedades colágenas,
infecciones, drogas, neumonitis por hipersensibilidad, etc.,
por lo cual su diagnóstico diferencial es complejo.
Su identificación es importante porque responde bien
a tratamiento corticoidal prolongado.
SARCOIDOSIS PULMONAR
La sarcoidosis es otra enfermedad intersticial
de causa desconocida que conviene tener presente porque no es
excesivamente rara y responde a corticoides, cuando no regresa
espontáneamente. Esta enfermedad compromete preferentemente
el pulmón y los ganglios hiliares y mediastínicos.
Con menor frecuencia afecta a los ojos, piel, sistema nervioso
e hígado.
Si bien su etiología es desconocida,
se ha planteado que el agente causal pudiera ser inhalatorio,
debido a la gran frecuencia del compromiso pulmonar. La reacción
del organismo es de tipo hipersensibilidad celular, que conduce
a la formación de granulomas y al aumento de linfocitos
T activados en el pulmón.
Anatomía patológica
Compromete especialmente el intersticio axial
peribronquial y la lesión característica es
el granuloma sarcoideo, parecido al tuberculoso, pero que
no evoluciona a la caseificación. Es de tamaño
microscópico y por confluencia forma nódulos
de color amarillo grisáceo. Microscópicamente,
está formado por células epitelioideas con abundante
citoplasma eosinófilo, linfocitos, células gigantes
multinucleadas con inclusiones, llamadas cuerpos asteroides
y de Schaumann. El mismo tipo de reacción histológica
puede encontrarse ocasionalmente en ganglios que drenan lesiones
TBC, neoplásicas, etc., hablándose en estos
casos de reacción sarcoide. Esta eventualidad debe
tenerse presente para la valoración de biopsias de
ganglios linfáticos.
Las lesiones suelen regresar espontáneamente
o bajo tratamiento, progresando en algunos casos hacia una
fibrosis terminal.
Evolución
Se distinguen tres formas evolutivas:
- Aguda: de regresión espontánea
antes de 2 años (78%).
- Prolongada: curso de 2 a 7 años,
también con regresión espontánea (14%).
- Crónica: con más de 7 años
de evolución, conduce a fibrosis pulmonar y muerte
por insuficiencia respiratoria, con o sin corazón
pulmonar crónico (8%).
La TBC es más frecuente en portadores
de sarcoidosis que en el resto de la población. También
suele observarse litiasis renal por calciuria.
Manifestaciones clínicas
El compromiso pulmonar de la sarcoidosis
es frecuentemente un hallazgo radiográfico en un paciente
asintomático. Cuando hay síntomas, éstos
son inespecíficos (fiebre, tos, disnea) y de intensidad
variable. El examen físico pulmonar es usualmente negativo.
La radiografía de tórax es fundamental para
la orientación diagnóstica inicial. Existen
diversas clasificaciones de los hallazgos radiológicos,
pero lo esencial es la presencia de adenopatías hiliares
o paratraqueales y compromiso intersticial. Ambos tipos de
lesiones pueden coexistir o presentarse aisladamente. En un
paciente joven y asintomático la imagen de adenopatías
hiliares bilaterales es fuertemente sugerente de esta condición
(Figura 40-1).
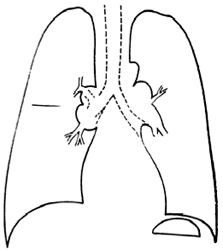
Figura 40-1. Sarcoidosis.
Las imágenes más características
son las adenopatías luiliares bilaterales, que
pueden o no acompañarse de compromiso pulmonar
reticular.
Con alguna frecuencia existe el antecedente
de eritema nodoso en las extremidades inferiores. En la sangre
suele encontrarse, al revés de lo que sucede en el
pulmón, una disminución de linfocitos T y un
aumento de los B, con hipergamaglobulinemia. Frecuentemente
aumenta la enzima convertidora de la angiotensina, pero esta
alteración no es específica. Puede existir hipercalcemia
y, posiblemente por la disminución de linfocitos T
fuera del pulmón, el PPD y otras reacciones de inmunidad
celular son negativos.
En lesiones focales las alteraciones funcionales
pueden no ser detectables y, en las formas más difusas
o que llegan a la fibrosis, se observan alteraciones restrictivas
e hipoxemia.
Diagnóstico
Ningún elemento, ni siquiera la histología,
es suficientemente específico como para fundamentar
el diagnóstico por sí solo. Sin embargo, por
la suma de varios de ellos y por la exclusión de otras
enfermedades es posible llegar al diagnóstico. La TAC
es particularmente valiosa para la detección de ganglios
y del compromiso del intersticio axial. Las biopsias bronquial
y transbronquial tienen un buen rendimiento diagnóstico.
La reacción de Kveim, por inyección intradérmica
de un extracto de nódulos sarcoidóticos, fue
en un tiempo el eje del diagnóstico, pero su uso prácticamente
se ha abandonado por la dificultad para obtener extractos
confiables.
Por su tendencia a la regresión espontánea,
el tratamiento con corticoides sólo se plantea en casos
muy sintomáticos o en aquellos con compromiso ocular
o neurológico.
|
|
|
